Primers:
| 1660 | Flk_GRB2_FWD | TCAGAACTGGTTCAAAGCTGAGT | JH | 6/1/2015 | 23 | 60 | O.lurida | GRB2 Flanking | P62994 | |
| 1659 | Flk_GRB2_REV | ACTGCGCTGACATACTGGAC | JH | 6/1/2015 | 20 | 60 | O.lurida | P62994 | ||
| 1658 | Flk_H3.3_FWD | CCAATGACAAATGAGCCACACAA | JH | 6/1/2015 | 23 | 60 | O.lurida | H3.3 Flanking | Q6P823 | |
| 1657 | Flk_H3.3_REV | TCGTACAAAGCAAACTGCACG | JH | 6/1/2015 | 21 | 60 | O.lurida | Q6P823 | ||
| 1656 | Flk_H2A.V_FWD | GCGATGGAGTTGATGAGGTG | JH | 6/1/2015 | 20 | 59 | O.lurida | H2A.V Flanking | P08991 | |
| 1655 | Flk_H2A.V_REV | CAAGGCAGTTTCTCGTTCGG | JH | 6/1/2015 | 20 | 59 | O.lurida | P08991 | ||
| 1652 | Flk_p29ING_FWD | GTGGACACACATGCACTCCT | JH | 6/1/2015 | 20 | 60 | O.lurida | p29ING4 Flanking | Q8C0D7 | |
| 1651 | Flk_p29ING_REV | AAGCAGACTCAGATTCAGGC | JH | 6/1/2015 | 20 | 58 | O.lurida | Q8C0D7 |
Reagent Table:
| Reaction_Components | Volume | Final Concentration |
| 2x Apex Red | 12.5 | 125 |
| Forward Primer (10uM) | 0.5 | 5 |
| Reverse Primer (10uM) | 0.5 | 5 |
| H20 | 10.5 | 105 |
| Template | 1 |
Using the final concentration I mixed each master mix going from largest to smallest volume.
I then pipetted 24 ul in each well followed by 1 ul of water or sample depending.
Then I ran the following PCR program:
| Temp | Time |
| 95 C | 5 min |
| 95 C | 30 sec |
| 55 C | 30 sec |
| 72 C | 30 sec |
| repeat steps 2-4 40 times | |
| 72 C | 3 min |
| 4 C | Hold |
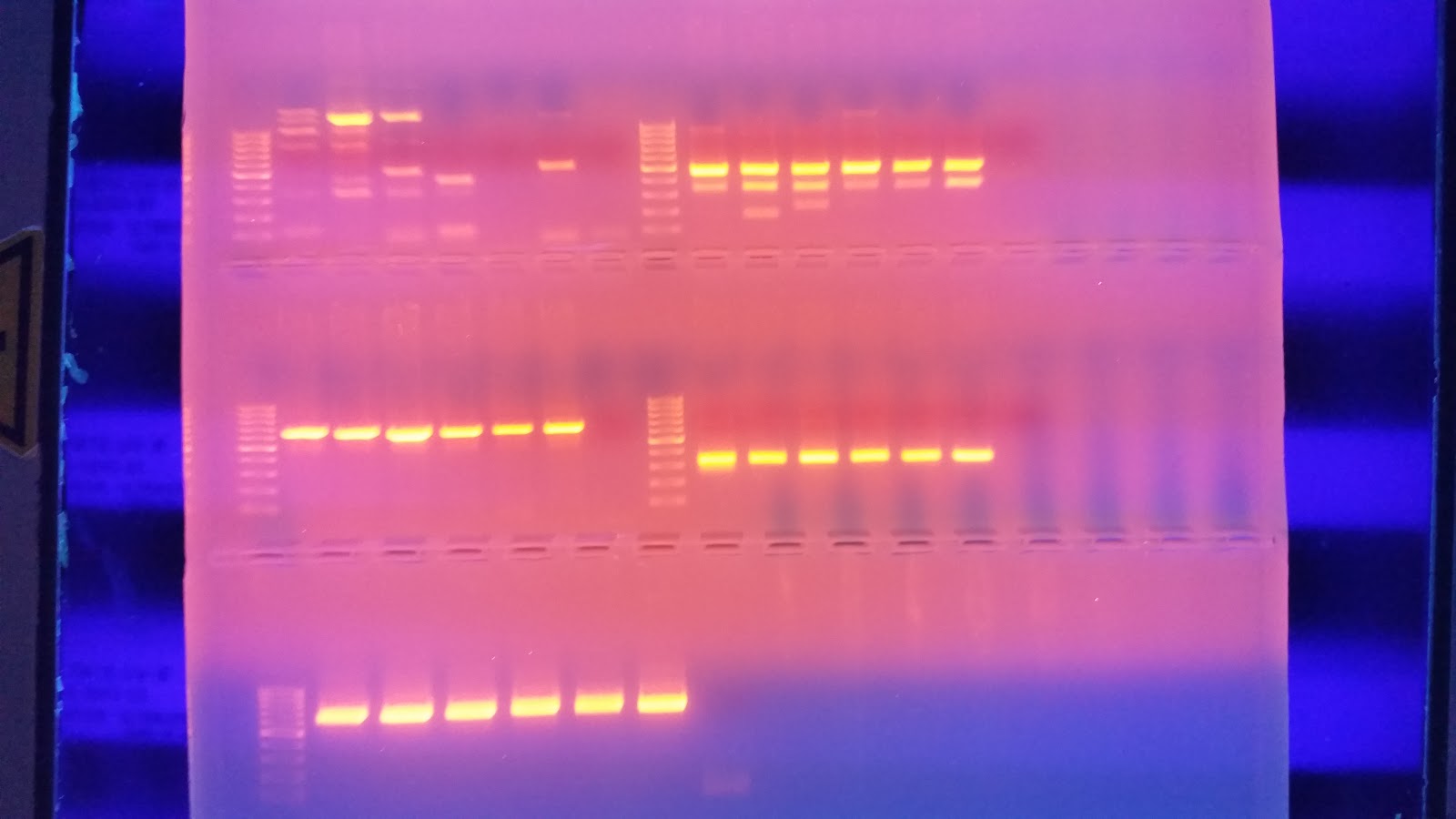
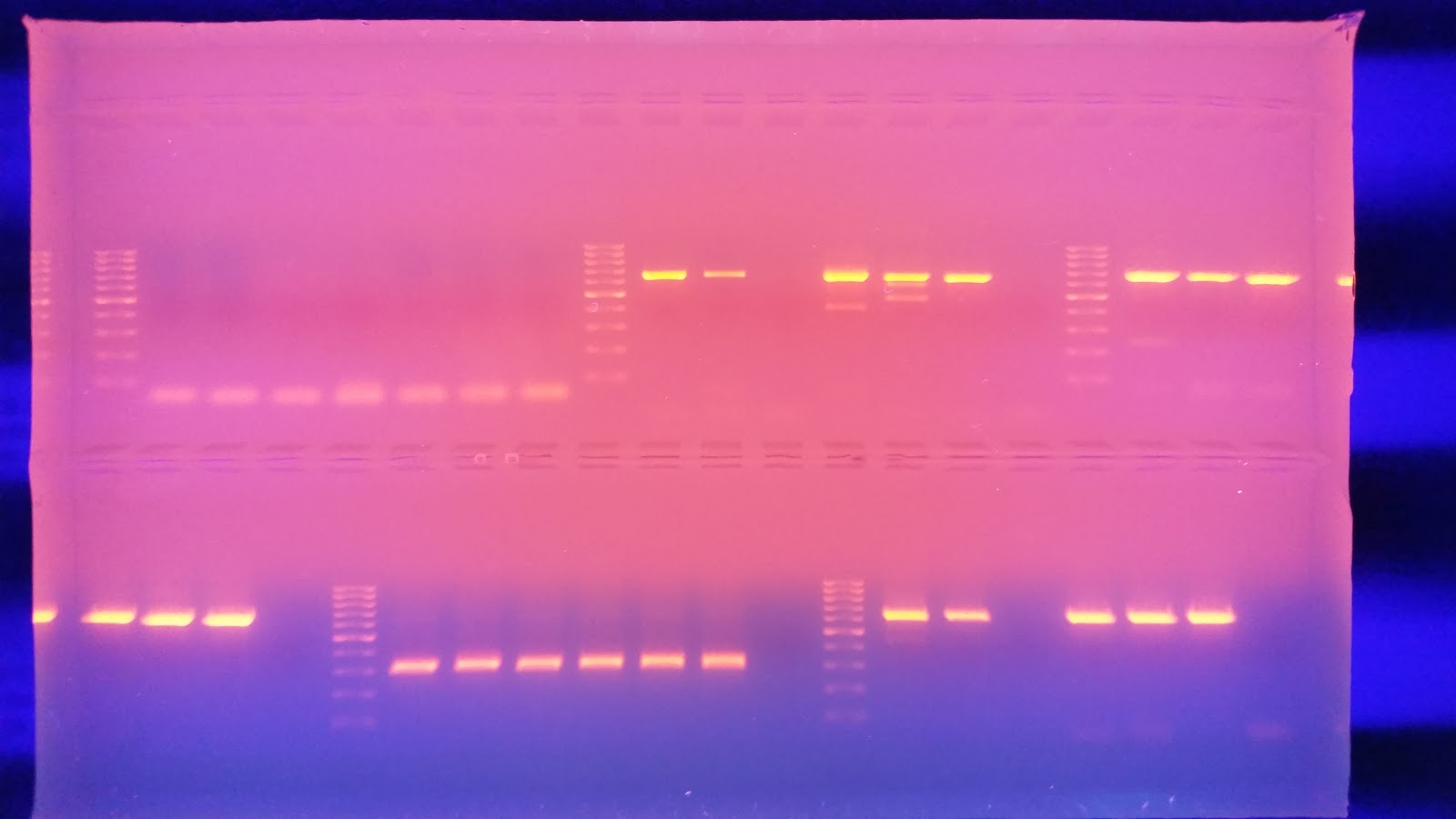
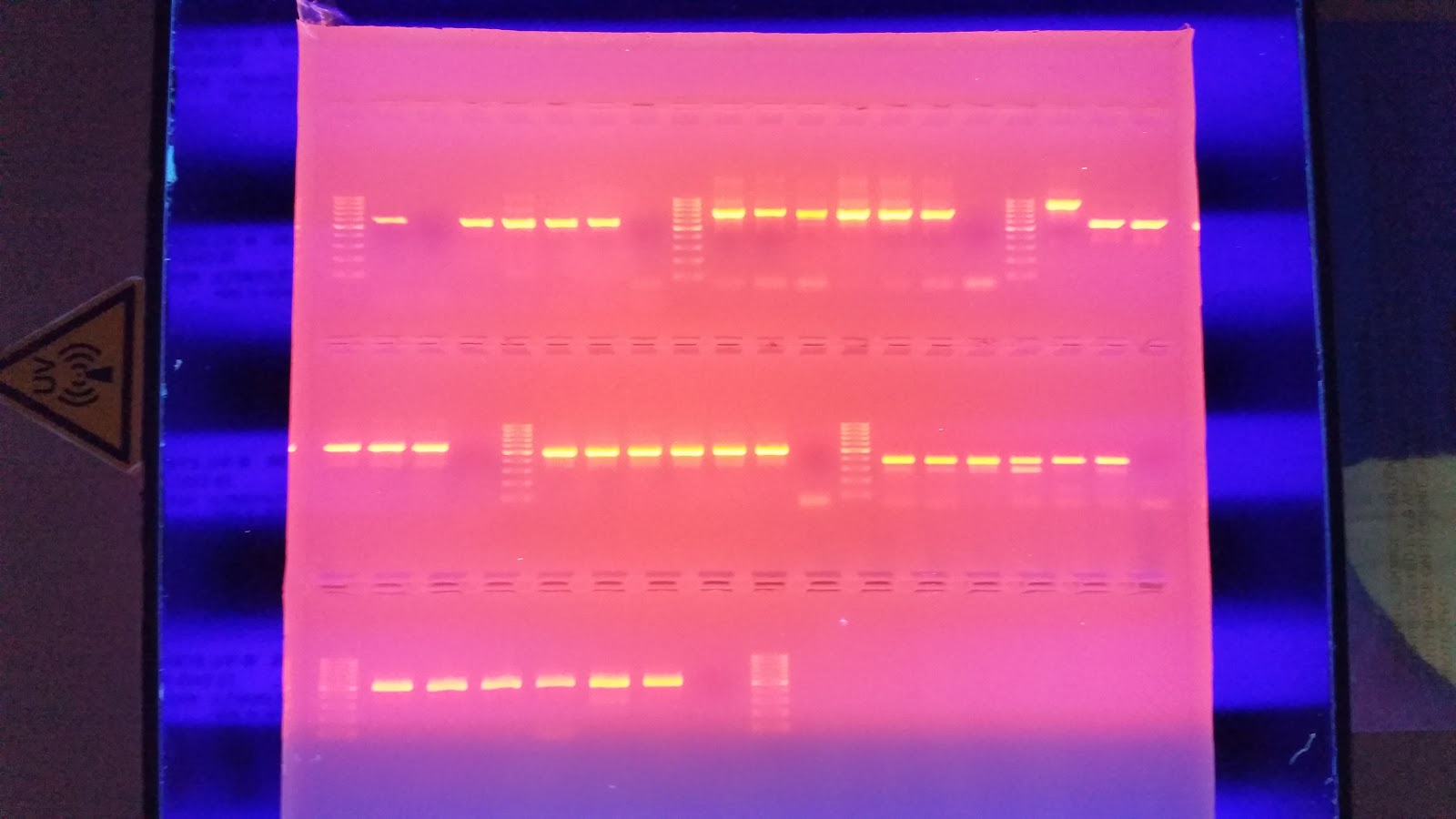
Once the PCR finished I ran 25 ul of each product on a 1.3% agarose gel
Gel Reagent Table
| Reagent | Volume |
| 1X Low TAE | 175 ml |
| Agarose | 2.3 g |
| EtBr | 17.5 ul |
- Add agarose to TAE.
- Microwave 1 minute stir
- Repeat until no particulate matter in solution
- Add EtBr while agarose still hot
- Gently pour in one corner of the gel cast until tray is full
I then ran the gel at 100 v for 40 minutes. I placed it on the transilluminator to view any bands that may have formed. In my haste I forgot to image the gel, Luckily the gel ran fine with strong banding and minor streaking due to over filled wells.
After running the gel I excised the bands. Once the bands were cut out of the gel and placed in a Millipore Purification Column and centrifuged at 5,000 rcf for 10 minutes to collect the purified DNA.
To send them off for sequencing I plated 4 replicates of 10 ul for each sample in the appropriate tube. On a second plate I plated two replicates of 3 uM primer working stock for each appropriate sample. These plates are now being held in the 4 C in 213 for sequencing.